




Scientific Papers in SCI
2023
2023
Materiales y Procesos Catalíticos de Interés Ambiental y Energético
Revealing the Impact of Different Iron-Based Precursors on the ‘Catalytic’ Graphitization for Synthesis of Anode Materials for Lithium Ion Batteries
Frankenstein, L; Glomb, P; Ramirez-Rico, J; Winter, M; Placke, T; Gomez-Martin, AChemelectrochem, 10 (2023) e202201073
Show abstract ▽
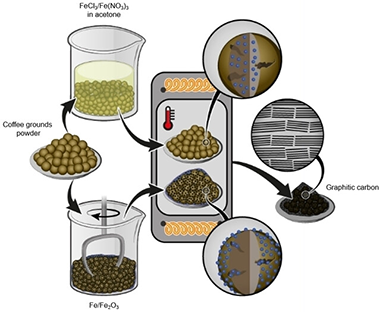
Low cost and environmentally friendly production of graphite anodes from naturally available biomass resources is of great importance to satisfy the increasing material demand for lithium ion batteries. Herein, graphitization of coffee ground was performed using four different iron-based activating additives, including iron (III) chloride, iron (III) nitrate, iron (III) oxide and pure iron, following either a wet or a dry mixing approach. The structural development regarding the type of activator used and the impact on the corresponding electrochemical performance are systematically investigated. A maximum degree of graphitization between 55 and 74 % (as determined by Raman spectroscopy) is attained using iron (III) chloride and iron powder, respectively. The graphitic anode material synthesized using iron powder reached a maximum reversible capacity of approximate to 320 mAh g(-1) at a rate of 0.1 C. This study provides significant insights into the impact of activators on the design of synthetic graphite from renewable sources.
| DOI: 10.1002/celc.202201073
2022
2022
Materiales Nanoestructurados y Microestructura
Pd supported on defective TiO2 polymorphic mixtures: Effect of metal-support interactions upon glycerol selective oxidation
Rinaudo, MG; Beltran, AM; Fernandez, A; Cadus, LE; Morales, MRResults in Engineering, 16 (2022) 100737
Show abstract ▽
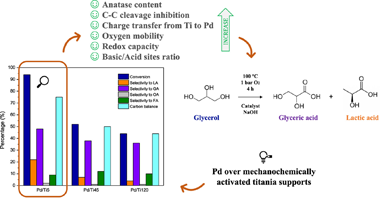
Palladium catalysts supported on defective mixes of anatase, TiO2 (II) and rutile crystalline phases, previously obtained by high-energy ball milling, were synthesized and tested for glycerol selective oxidation. A deep characterization of these unusual materials was carried out to elucidate catalytic and physicochemical features. Electron density transfer from support to metal or vice versa, depending on the polymorphs present, could not only alter palladium particle sizes and its surface oxidation state but also reducibility and oxygen mobility of catalysts. Furthermore, acid-base properties achieved also influenced catalytic activity under mild conditions of liquid-phase glycerol oxidation. A conversion of 94% and a selectivity to glyceric and lactic acids of 48% and 22% respectively were obtained for the Pd catalyst supported on mechanochemically activated anatase. The presence of several polymorphs in a metal oxide support could therefore benefit or handicap catalytic cycle for a particular reaction. Metal-support interactions play a key role in heterogenous catalysts and thus the rational design of supports comes on the scene.
December, 2022 | DOI: 10.1016/j.rineng.2022.100737
Reactividad de Sólidos
Theoretical Analysis of Polynuclear Zinc Complexes Isolobally Related to Hydrocarbons
Ayala, R; Galindo, AInternational Journal of Molecular Sciences, 23 (2022) 14858
Show abstract ▽
Based on the isolobal analogy of ZnCp (Cp = eta(5)-C5H5) and ZnR (R = alkyl or aryl group) fragments with hydrogen atom and fragment [Zn(CO)(2)] with a CH2 carbene, the following complexes [(ZnCp)(2){mu-Zn(CO)(2)}], 1, [(ZnPh)(2){mu-Zn(CO)(2)}], 2, [(ZnPh){mu-Zn(CO)(2)}(ZnCp)], 3, [(ZnCp)(2){mu-Zn-2(CO)(4)}], 4, [(ZnPh)(2){mu-Zn-2(CO)(4)}], 5, [(ZnPh){mu-Zn(CO)(2)}(2)(ZnCp)], 6, [Zn-3(CO)(6)], 7 and [Zn-5(CO)(10)], 8, were built. These polynuclear zinc compounds are isolobally related to simple hydrocarbons (methane, ethane, cyclopropane and cyclopentane). They have been studied by density functional theory (DFT) and quantum theory of atoms in molecules (QTAIM) to compare the nature and topology of the Zn-Zn bond with previous studies. There are bond critical points (BCPs) between each pair of adjacent Zn centers in complexes 1-8 with Zn-Zn distances within the range 2.37-2.50 angstrom. The nature of the Zn-Zn bond in these complexes can be described as polar rather than pure covalent bonds. Although in a subtle way, the presence of different ligands and zinc oxidation states introduces asymmetry and polarity in the Zn-Zn bond. In addition, the Zn-Zn bond is delocalized in nature in complex 7 whereas it can be described as a localized bond for the remaining zinc complexes here studied.
December, 2022 | DOI: 10.3390/ijms232314858
Química de Superficies y Catálisis
Materials challenges and opportunities to address growing micro/ nanoplastics pollution: a review of thermochemical upcycling
Parrilla-Lahoz, S; Mahebadevan, S; Kauta, M; Zambrano, MC; Pawlak, JJ; Venditti, RA; Reina, TR; Duyar, MSMaterials Today Sustainability, 20 (2022) 100200
Show abstract ▽
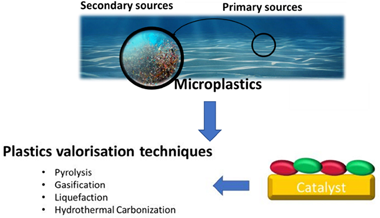
Micro/nanoplastics have sparked attention in recent years due to their widespread presence in the environment. Currently, several waste valorization approaches are under development in order to upcycle micro/nanoplastics. Thermal conversion technologies such as pyrolysis, gasification, liquefaction, or hydrothermal carbonization can yield high-value solid products, oil, and gases from plastics waste. The common thermal conversion technologies investigated focus on maximizing the production of oil and gases (such as H2 and CH4) for use as fuel. Except for hydrogen, when these products are used to generate energy, the carbon emissions generated are comparable to those produced by traditional fossil fuels. Herein, we present a review of the current efforts to capture and convert plastic waste into valuable products with an emphasis on identifying the need to develop processes specifically for micro/nano-plastics while also preventing the release of CO2 emissions. We identify the development of efficient catalytic materials as a critical research need for achieving economically viable thermochemical con-version of micro/nanoplastics.
December, 2022 | DOI: 10.1016/j.mtsust.2022.100200
Química de Superficies y Catálisis
Development of Power-to-X Catalytic Processes for CO2 Valorisation: From the Molecular Level to the Reactor Architecture
Bobadilla, LF; Azancot, L; Luque-Alvarez, LA; Torres-Sempere, G; Gonzalez-Castano, M; Pastor-Perez, L; Ramírez-Reina, T; Ivanova, S; Centeno, MA; Odriozola, JAChemistry, 4 (2022) 1250-1280
Show abstract ▽
Nowadays, global climate change is likely the most compelling problem mankind is facing. In this scenario, decarbonisation of the chemical industry is one of the global challenges that the scientific community needs to address in the immediate future. Catalysis and catalytic processes are called to play a decisive role in the transition to a more sustainable and low-carbon future. This critical review analyses the unique advantages of structured reactors (isothermicity, a wide range of residence times availability, complex geometries) with the multifunctional design of efficient catalysts to synthesise chemicals using CO2 and renewable H-2 in a Power-to-X (PTX) strategy. Fine-chemistry synthetic methods and advanced in situ/operando techniques are essential to elucidate the changes of the catalysts during the studied reaction, thus gathering fundamental information about the active species and reaction mechanisms. Such information becomes crucial to refine the catalyst's formulation and boost the reaction's performance. On the other hand, reactors architecture allows flow pattern and temperature control, the management of strong thermal effects and the incorporation of specifically designed materials as catalytically active phases are expected to significantly contribute to the advance in the valorisation of CO2 in the form of high added-value products. From a general perspective, this paper aims to update the state of the art in Carbon Capture and Utilisation (CCU) and PTX concepts with emphasis on processes involving the transformation of CO2 into targeted fuels and platform chemicals, combining innovation from the point of view of both structured reactor design and multifunctional catalysts development.
December, 2022 | DOI: 10.3390/chemistry4040083
- ‹ previous
- 36 of 410
- next ›


![]()
icms



